What is iTP-Seq?#
Uneven translation rates resulting from mRNA context, tRNA abundance, nascent amino acid sequence or various external factors play a key role in controlling the expression level, regulation and folding of the proteome.
Inverse toeprinting coupled to next-generation sequencing (iTP-Seq) [1] is a scalable in vitro method for characterizing bacterial translation landscapes, complementary to ribosome profiling (Ribo-Seq) [2] [3], a widely used method for determining transcriptome-wide protein synthesis rates in vivo.
In iTP-Seq, ribosome-protected mRNA fragments known as inverse toeprints are generated using RNase R, a highly processive 3’ to 5’ RNA exonuclease. Deep sequencing of these fragments reveals the position of the leading ribosome on each mRNA with codon resolution, as well as the full coding regions translated by these ribosomes.
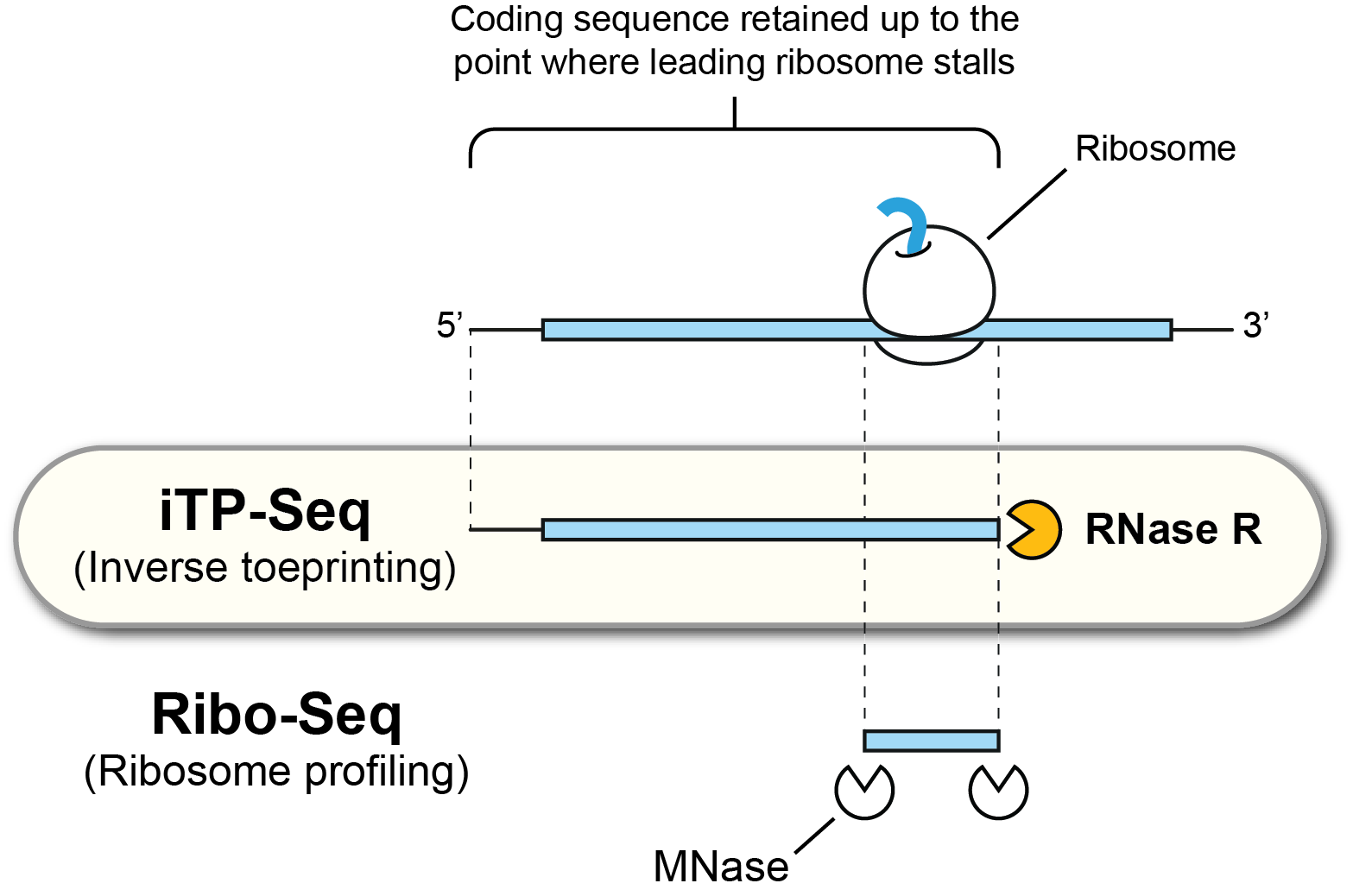
Figure 1. Overview of the iTP-Seq methodology.#
Unlike Ribo-Seq, in which short mRNA footprints are mapped to a sequenced genome, no a priori knowledge of the translated sequences is required for iTP-Seq, making it possible to work with fully customizable transcript libraries. As a standardized framework for inverse toeprint generation, amplification and sequencing, iTP-Seq can therefore be used in combination with different types of libraries, in vitro translation conditions and data analysis pipelines tailored to address a range of biological questions involving context-dependent translational arrest, as exemplified by our previous studies on ribosome-targeting antibiotics [4] [5].
A detailed experimental protocol for performing iTP-Seq can be found here and is outlined below.
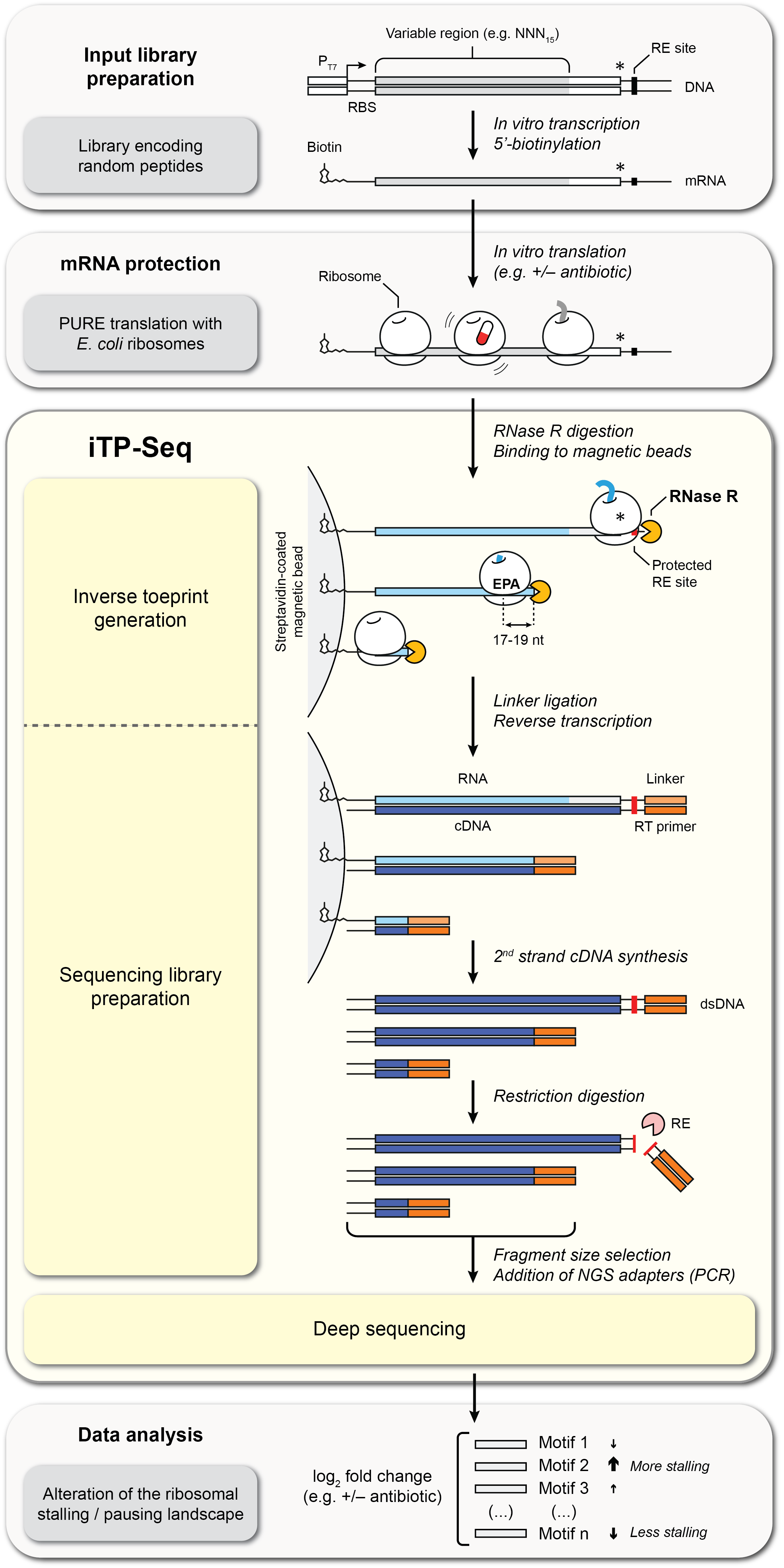
Figure 2. Experimental workflow for iTP-Seq analysis.#
Bioinformatic analysis of the resulting iTP-seq data can be performed using the itpseq software available on this website, as explained in our various tutorials.
References: